
Zeba Hasan Hafeez ( Departments of Dermatology, Civil Hospital. Karachi. )
Tahir Hussain ( Departments of Medicine, Civil Hospital, Karachi. )
September 1996, Volume 46, Issue 9
Case Reports
Introduction
Lipoid protcinosis is a rare, autosornal recessive disorder, characterized by widespread deposits of hyalinc material in the skin, oropharynx, larynx, other mucosal surfaces and various internal organs. Only about 200 cases have been described in literature. As far as the author is aware, this is the first family being reported from Pakistan.
Case Report
Case 1
A twelve year old boy from Tando Adam (Sindh. Pakistan) presented with hoarseness, generalized blistering including the oral mucosa and itching which started when he was five months old. At one year, he began to have difficulty in opening his mouth, protrusion of tongue and feeding. Intermittent abdominal pain associated with nausea and vomiting, began at seven years. A fit followed by unconsciousness affected the right half of the body and face, one and a half years ago. His parents were first cousins. Patient is the second child often siblings. Three siblings died of epilepsy during infancy. Lipoid proteinosis is present in seventh female and tenth male siblings. Aunt is similarly affected.
On examination, his skin over the face, ears, back of neck, arms, trunk, buttocks was thickened and waxy with areas of scarring. Yellowish, waxy plaques were seen in axillac, elbows, groin and base of penis. Two erosions were present on the abdomen and nose. Shins were xerotic. teeth mal-aligned, tongue enlarged with restricted movements. Oral
mucosa was whitish and thickened. A few small hyperkeratotic plaques were seen on the back of hands. Scalp was thickened, scaly with scattered crusts. Beaded perifollicular papules were present on eyelids; cornea and fundi clear. Liver was 3 cm enlarged.
The investigations showed basic hematology and biochemistry as normal. Serum cholesterol was 95 rng% (N 140-220). triglycerides 58 rng% (N 140-220). HDL 29 mg% (N more than 35), LDL 54 rng% (N upto 150) and phospholipids 200 rng% (N 125-135). Serum proteins showed a low albumin and etectrophoresis revealed increased alpha 2, gamma and to a lesserextentbeta globulins. Thickening of the right and left vocal cords and anterior commissure were observed at direct laryngoscopy. TJppcr G.!. endoscopy was normal. Stomach and duodenal biopsies were consistent with chronic superficial gastntis and duodenitis. Ultrasound showed liver to be slightly enlarged with impression of fatty infiltration. EEG revealed a normal background activity with few single spikes in the right frontal regions. MRI of brain was normal.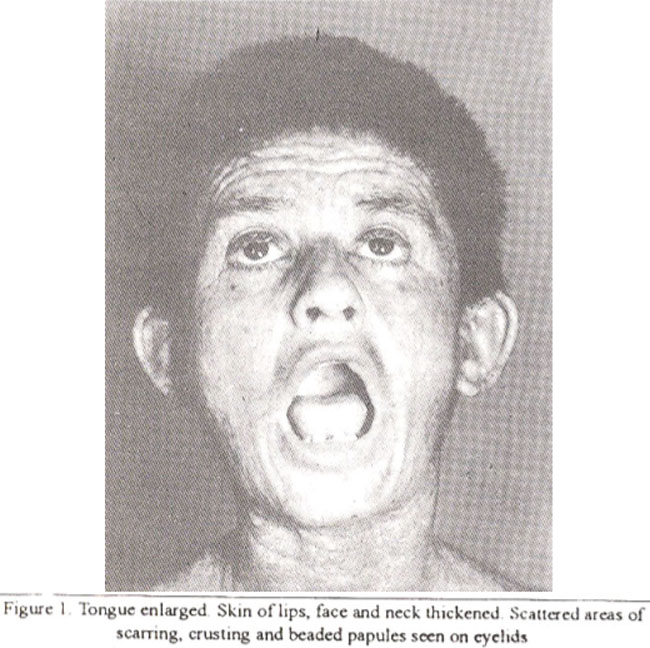
Liver biopsy showed intact lobular architecture and a few inflammatory cells. Some cells had lipid vacuoles. Portal areas were widened with fibrosis and infiltrated by inflammatoly cells. A few vessels at the margins had hyaline thickening of the walls. Skin biopsy of the axilla showed irregularly acanthotic epidermis: dermis was hyalinized with dermal capillaries having thick, hyalinized walls and narrowed lumenae. A similar hyalinized band was seen around sweat glands. These hyalinized areas were PAS positive and diastase resistant. Oral mucosal biopsy revealed stratified squamous epithelium overlying homogenized conium, staining positively with PAS. Blood vessels were thickened and surrounded by a similar PAS positive material.
Case 2
Patient’s eight years old aunt developed hoarseness, generalized and oral mucosal blistering at age six months and abdominal pain with vomiting off and on at the age of two ears. Pruritus was also present. On examination, a few patches of scarring alopecia, beaded yellowish papules on eyelids and below eyebrows were seen. Skin of face, buttocks and elbows was thickened and waxy with a few scars. Hyperkeratotic plaques were seen on elbows and back of the hands. Oral mucosa and tongue were thickened. Liver was 3 cm enlarged. Skin and oral mucosal biopsies were suggestive of lipoid proteinosis. Chronic superficial gastritis and duodenitis were seen on endoscopy.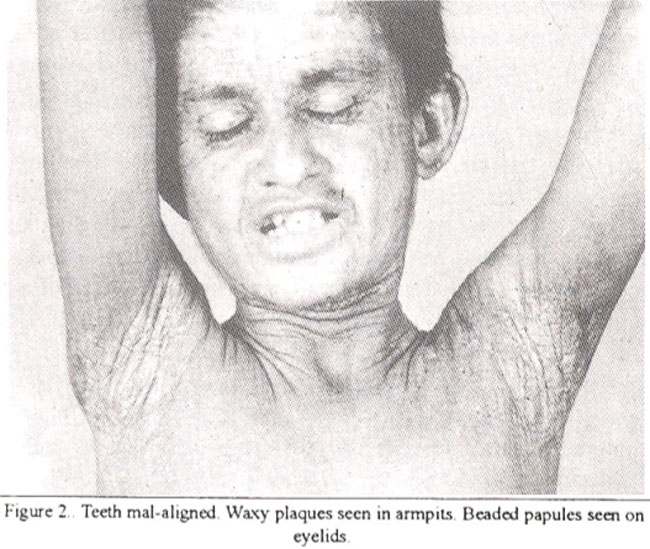
Case 3
Patient’s seventh female sibling aged 2-1/2 years had hoarseness, thickening and difficulty in protruding her tongue from the age of one month. Generalized and oral mucosal blistering began at six months of age. On examination. hypopigmented macules were seen on trunk. Bullae and erosions were present on elbows, axillae and trunk. Milestones appeared to be delayed.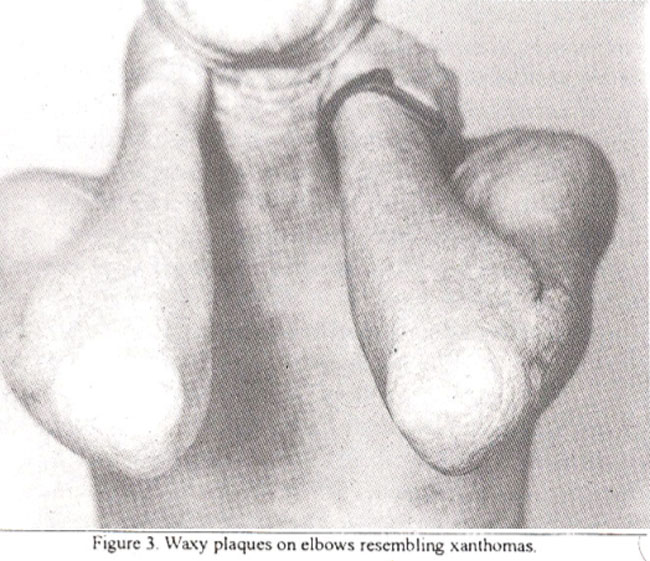
Case 4
Patient’s tenth male sibling aged seven months had hoarseness, blisters and pustules on scalp from age three months. At seven months, he had severe pneumonia followed by generalized and oral mucosal blistering.
Discussion
Lipoid proteinosis cutis et mucosae (Urbach-Wiethc disease) is a recessively inherited, multisystemic disorder primarily affecting the skin, oral cavity and laiynx. It runs a chronic, benign course. Although it causes functional and cosmetic problems, the mortality is low, except in severe laryngeal obstniction.
About 200 cases have been reported in literature, with increased incidence in South Africa and Sweden1. Both sexes are equally affected. The first case was reported by Siebenman (1908) as “generalized hyperkeratosis of the skin with involvement of the mucosae"2. Lutz and Rossle designated similar cases as "nevus ichthyosiformis” and "hereditary progressive pachydermic dystrophy”2. Wiethe reported six cases. Urbach stated that the disease was a disturbance of lipid metabolism and named it “lipoidosis cutis et mucosae”. He subsequently changed this appellation to “lipoidproteinosis"2. It was also thought to be a lysosomal storage disease3, or a primary disturbance of collagen metabolism4. More recently, ultrastructural and phenotypic changes in the fibroblasts of lipoid proteinosis have been identified which may be the cause of pathological changes5.
Histologically, the epidermis is hyperkeratotic with irregular acanthosis. The dermis is thickened. PAS positive hvaline material is deposited around capillaries and sweat glands. Ultrastructurally, hyaline material is interspaced betweencollagen bundles. Accumulationof type four and five collagen is seen around blood vessels and appendages. Type three collagen is abnormally distributed, with reduction of type one collagen1.
Clinically, symptoms begin early in life. Inflammatory lesions i.e., bullae and pustules are followed by slow healing, acneform, varioliform or papulonecrotic scars affecting the face and limbs6. Hoarseness begins early due to inelasticity of vocal cords and becomes prominent with time. Yellowish white infiltrates develop in the phaiynx, tongue and lips. Gingivat infiltration9 has been reported recently. The tongue is enlarged, firm and movements are diminished. Occasionally. mucosae of labia and vagina are similarly affected. Pruntus, tendency to bruise easily and regional hyperkeratosis are prominent secondary signs. Yellowish brown nodules appear on the face and limbs. Those on the limbs resemble xanthomas. ‘Beaded’ papules develop along margins of eyelids. Loss of eyelashes and cicatricial alopecia, may result.
Dental abnormalities, recurrent parotiditis, epilepsy and intracranial calcifications10 (hippocampal6 and temporat11,12) have been reported. Xerostornia associated with slight keratoconjunctivitis sicca;13 and eye changes (dejosilion of hyalinoid substance in the retina). corectopia14 and pseudomembranous conjunctivitis15 have been seen. Widespread visceral involvement16 with virtually every organ has been described. Familial incidence of lipoid proteinosis17-20 has been reported in literature.
Disturbance of serum proteins (increase of alpha 2 and gamma globins and to a lesser extent beta globulins)2 is seen with low albumin. Alimentary glycosuria and abnormal glucose tolerance test2 have also been reported. In the treatment, surgical removal of vocal cord infiltrates has been suggested. Tracheostomy is necessaiy in some cases of laryngeal obstruction. Dermabrasion, chemical skin peeling, blepharoplasty, DMSO21 and etretinate22 have been tried.
This patient had almost all the features of lipoid proteinosis except for visceral involvement. He was treated with etretinate, one mg per kg body weight for three months. However, there was no improvement of skin lesions, pruritus and hoarseness (no changes at direct laiyngoscopy). The patient was lost to follow-up.
References
1. Harper. J.I., Duance, V.C., Sims, T.J. et at. Lipoid protcinosis: An inherited disorder of collagen metabolism? Br. J. Dermatol., 1985;113:145-51.
2. Laymen, C.W. and Hill, EM. An appraisal of hyalinosis cutis ct mucosac. Arch. Dcrmatol., 1957;75:55-65.
3. Bauer, E.A., Santa-Cruz, Di. and Eison, A.Z. Lipoid proteinosis: In vivo and in vitro evidence for a lysosomal storage disease. J. Invest. Dermatol., 1981;76:119-25.
4. Harper, J.I., Filipe, Ml. and Staughton, R.C.D. Lipoidproteinosis: Variations in thehistochemical characteristics. Clin. Exp. Dermatol., 1983;8:135-41.
5. Moy, L.S., Moy, R.L., Matsuoka, L.Y. et at. Lipoid proteinosis: Ultrastructural and biochemical studies. J. Am. Acad. Dermatol., 1987;16:1193-201.
6. George, HF. Lipoid proteinosis. Derm in Gen Medicine. Thomas B Fitzepatrick, Arthur Z Eisen, Klaus Wolff, Irwin M Freedberg, Frank Austen Third edition, New York, McGraw-Hill Inc., 1987, pp. 1760-63.
7. Harper, 3.1. Oropharyngeal and larygeal lesions in lipoid proteinosis. J. Laryngol. Otol., 1983;97:877.
8. Seth, L.S.. Bhargava, S.K.. Dandekar, K.N. et at. Lipoid prroteinosis. Pak. J. Otolaryngol., 1993;9:203-5.
9. Israel, H. Gingival lesions in lipoid protcinosis. J. Peridontol., 1992;63;561-4.
10. Black, M.M Lipoid protcinosis. Text book of dermatology Rook, Wilkingson, Elbing, Fifth edition, London, Black well Scientific Publications, 1992, pp. 2347-85.
11. Hardcastle, SW. and Rosenstrauch, W.J. Lipoid proteinosis. A case report S. Aft. Med. J., 1984;66:273-4.
12. Emsley, R.A. and Paster, L. Lipoid proteinosis presenting with neuropsychiatric manifestations. J. NeuroL Neurosurg. Psychiatry, 1985;48: 1290.
13. Disdier, P., Harle, JR., Andrac, I. et al Specific xerostomia during UrabchWiethe disease. Dermatology, 1994; 188:50-1.
14. Johnson, L.N. and Helper, R.S. Corectopia and lipoid protcinosis. Br. J. Opthalmol., 1989,73:394-6.
15. Barthelemy, 3.. Mauduit, G., Kanitakis, J. et at. Lipoid proteinosis with pscudomanbranous conjunctivitis. J. Am. Acad. Dermatol., 1986;14 (Pt 2):367-71.
16. Caplan. R.M. Visceral involvement in lipoid proteinosis. Arch. Dermatol., 1967;95:145-55.
17. Chularatna, W., Harendra-de-Silva, D.G., Ruberu, R. et al. A Sri Lankan family with lipoid proteinosis (letter). Ceylon Med. J., 1995;40:50-1.
18. Ozbek, S.S.. Akyar, S. and Turgay, M. Case report: Computed tomography findings in lipoid proteinosis; Report oftwo cases. Br. J. Radiol., 1994,67:207-9.
19. Oezarmagan. 0.. Baykal, C. and Gursoy. E.O. Lipoid protcinosis in two sisters. Hautarzt, 1993;44:315-8.
20. Gruntzig, J. Hyalinosis cutis et mucosae (lipoid proteinosis Urbach-Wiethe) Fortschr. Med., 1983; 101 :690-6.
21. Wong, C.K. and Lin, CS. Remarkable response of lipoid proteinosis to oral dimethyl sulphoxide Br. J. Dermatol., 1988;119:541-4.
22. Dowlati, A., Dowlati, V., Mansauri, P. et al. Lipoid proteinosis and its response to ctretinate therapy. In: Pierard G.E.. Pierard-Franchiomont, C. eds. The Dermis: From Biology to disease. Paris, Monographies Dermatopathologiques Liegoises, 1989, pp. 135.42.
Related Articles
Journal of the Pakistan Medical Association has agreed to receive and publish manuscripts in accordance with the principles of the following committees: