
Saqib Qayyum Ahmad ( Department of Pathology, PAF Hospital, Mianwali. )
Mudassar Iqbal ( Department of Paediatrics, PAF Hospital, Mianwali. )
Madiha Saeed Wahla ( Department of Radiology, PAF Hospital, Mianwali. )
Aimel Munir Tarrar ( Department of Surgery, PAF Hospital, Mianwali. )
November 2011, Volume 61, Issue 11
Case Reports
Abstract
Thalassaemia intermedia includes thalassaemias with clinical severity intermediate between asymptomatic thalassaemia minor and transfusion dependent thalassaemia major. By definition patients of thalassaemia intermedia maintain a haemoglobin level of 7-10 g/dl and do not, or only occasionally, require blood transfusion. An eight-year-old girl who was a known case of thalassaemia intermedia and had been occasionally transfused presented with fever, pain and swelling over the wrists, ankles and above the right knee joint. Radiographs showed medullary widening, cortical thinning and; multiple, recent and old, partially healed fractures of metadiaphseal regions of long bones. Her fractures have been immobilized by means of back slabs. In view of her recurrent fractures and growth retardation we advised a regular transfusion-chelation regimen to our patient to suppress her ineffective dyserythropoiesis. The treatment is expected to prevent further bone fragility and fractures, as well as improve her life quality.
Keywords: Thalassaemia intermedia, Transfusion-chelation, Fractures.
Introduction
Thalassaemia intermedia (TI) includes patients of thalassaemia with clinical severity intermediate between asymptomatic thalassaemia minor and transfusion dependent thalassaemia major (TM). Patients of TI maintain a haemoglobin level of 7-10 g/dl and do not, or only occasionally, require blood transfusion.1 There are many genetic causes of TI but most of the cases are due to globin gene mutations.2 Severe cases of TI present with some unique clinical problems such as bone deformities and fractures. These problems are caused by multiple factors but mainly because of erythroid expansion due to ineffective erythropoiesis, and resultant cortical thinning.2 Since there are no definite guidelines, treatment often has to be tailored for individual cases.1 A case of TI with multiple fractures is presented followed by discussion on management options.
Case Report
An eight-year-old girl, a known case of thalassaemia intermedia, presented with fever and pain over ankles and wrists of one week duration; and two days\\\' history of pain over right thigh with inability to get up and stand on her feet. There was no history of trauma. She had been diagnosed at the age of two years, as suffering from homozygous thalassaemia on Hb electrophoresis, which had shown 97% foetal haemoglobin (HbF). Mutation analysis showed homozygous, codon 30(G-C) mutation. She had never received blood transfusion until one year ago and since then she has been transfused four times at various hospitals. Her parents are first cousins and she has two brothers and four sisters. All are healthy except a 3 years old younger brother who is also a case of TI.
On examination she appeared about 4 years of age and her weight (15 kg) and height (1.13 m) were below third percentile. She was pale and had swelling and tenderness over both wrists, both ankles and right thigh just above the knee joint. Her spleen was palpable 4 cm below left costal margin.
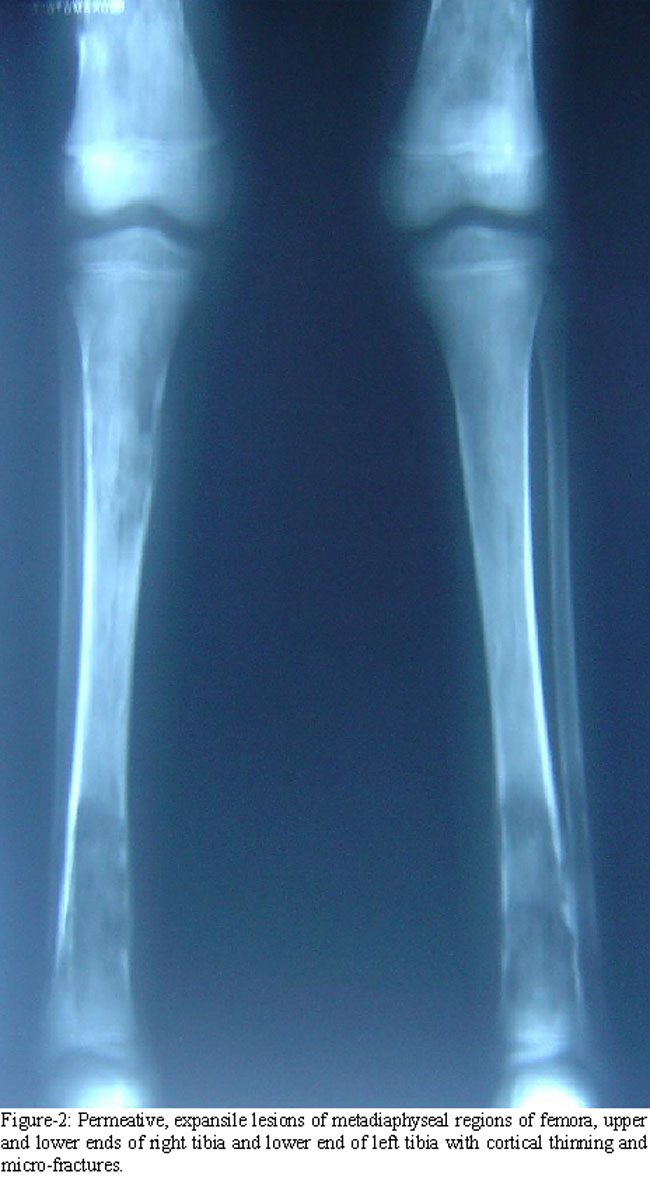
Her haemoglobin was 7.2 g/dl while white cell count and platelets\\\' count were 5.2x109/l and 210x109/l respectively. Blood smear showed a bizarre red cells\\\' morphology with marked anisopoikilocytosis, and a few nucleated red cells. Serum ferritin was 640 ng/ml. Blood sugar level; thyroid, liver and renal function tests were normal. She was negative for hepatitis screening. Radiological survey revealed multiple new and old permeative lesions causing marrow expansion, cortical thinning and microfractures in metadiaphyseal regions of long bones (Figure-1 and 2).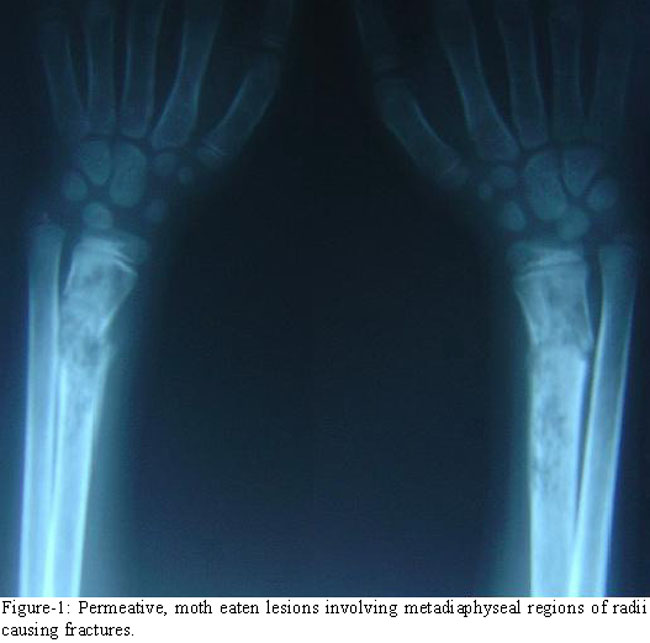
She was managed with intravenous broad-spectrum antibiotics and antipyretics along with folic acid, vitamin D, calcium and ascorbic acid supplements. Her fractures were splinted with back slabs. Keeping in view her recurrent multiple fractures it was decided to maintain her haemoglobin above 9 g/dl through a regular transfusion-chelation regimen. After a follow-up two months, patient has shown marked symptomatic improvement.
Discussion
There is a great variability in severity and clinical course of the patients with TI. At one end of the spectrum, there are the patients with mild TI having minimal symptoms; while at the other end we see patients of severe TI, with pronounced symptoms starting at an early age. The latter usually present at about one and half to three years of age, with features such as worsening anaemia, impaired growth, marrow hyperplasia, bony deformities, fractures and splenomegaly.3 They also later develop extramedullary haemopoiesis, iron overload, endocrinopathies, hypercoagulability, pulmonary hypertension, gall stones and require transfusion more often.4
One of the basic pathogenetic factors in thalassaemia syndromes is imbalance between and globin chains and severity of disease is generally proportional to the degree of imbalance. However because of various genetic and environmental modifiers, a single genotype can result in diverse clinical phenotypes.5 Our patient was found to be homozygous for Codon 30 (G-C) mutation, which is not among the common mutations seen in Pakistani population and does not necessarily produce the same phenotype in all the patients because of various genetic and environmental modifiers.6
Since the inception of hypertransfusion-chelation therapy about three decades ago, bony deformities and fractures are now rarely seen in well-managed patients of TM.7 On the other hand, these problems are still common in patients with severe TI. Expansion of haemopoietic marrow due to ineffective erythropoiesis causes mechanical interruption of bone formation and results in cortical thinning and fractures, as are also obvious in the radiographs of our patient (Figures-1 and 2). Later, over the course of illness, as the iron overload increases, endocrinopathies viz hypopituitarism, hypothyroidism, parathyroid dysfunction, growth hormone deficiency and hypogonadism develop and contribute to osteoporosis which causes further bone fragility.8
In our patient serum ferritin level is moderately raised even after eight years of illness and stigmata of iron overload induced endocrinopathies are not marked. Iron overload in TI occurs mainly due to increased absorption from the gut, and occurs more slowly as compared to iron overload caused by blood transfusions.8 In cases of TI, increased production of growth and differentiation factor 15 (GDF-15) by dyserythropoietic cells and increased expression of hypoxia inducible transcription factors (HIF) suppress Hepcidin production by the liver, which causes increased gut absorption of iron.8
Although stem cell transplantation is the best treatment for patients of TI but this option is unfortunately not available to most of the patients. On the other hand definite guidelines for the medical management of TI patients are also not available and treatment often has to be individualized.
Presently the general practice is to avoid blood transfusion in patients of TI, as much as possible, to prevent transfusion dependence. However most of the patients of TI do require random transfusions as they reach adolescence, with a frequency proportional to disease severity. Indications for transfusion include falling haemoglobin, bony changes, growth failure, pulmonary hypertension, aplastic crisis, thromboembolism and pregnancy.1,9 Aim of these random transfusions is palliation of the complications of TI.
A preventive approach is now also advocated in patients of severe TI and consists of regular transfusion-chelation therapy from the very young age in order to prevent the complications.9,10 The evidence in favour of benefits of regular transfusion-chelation can be drawn from the well managed cases of TM, where it has prevented or markedly reduced complications such as fractures, extramedullary haemopoiesis, endocrinopathies, thromboembolism, and pulmonary hypertension.7 The disadvantages include problems concerning regular availability of safe blood, inconvenience related to transfusion-chelation, iron overload, infections and alloimmunization. Initiation of regular transfusion at a later age is more commonly associated with alloimmunization than starting transfusion at an early age.7
We have advised regular blood transfusion-chelation to our patient due to repeated fractures and poor growth. The objective is to keep haemoglobin above 9.0 g/dl and suppress her own erythropoiesis, which is the main cause of her repeated fractures. If the bony changes are reversed, there will always be an option to give a trial of weaning her off from regular transfusion at a later stage.
Other treatment options include splenectomy and modulation of HbF. Splenectomy is indicated if patient has hypersplenism, increased transfusion demand or physical encumbrance.1,10 However recent evidence suggests that splenectomy is more commonly associated with complications such as hypercoagulability, infections and pulmonary hypertension.10 Trials with HbF modulators such as hydroxyurea, butyrate and its derivatives and thalidomide have shown only modest benefits.10 In our patient these options remain open for consideration as adjuvant therapy during the course of her illness.
Conclusion
There are no definite guidelines regarding medical treatment of TI. Treatment in most cases is tailored for each patient individually. Blood transfusion therapy must be considered in selected group of patients of TI after weighing benefits against the disadvantages.
References
1.Tahir A, Ismaeel H, Cappellini M D. Thalassaemia intermedia: Revisited. Blood Cells Mol Dis 2006; 37: 12-20.
2.Mohamed N, Jackson N. Severe thalassaemia intermedia: clinical problems in the absence of hypertransfusion. Blood Rev 1998; 12: 163-70.
3.Phadke SR, Agarwal S. Phenotype score to grade the severity of thalassemia intermediate. Indian J Pediatr 2003; 70: 477-81.
4.Borgna-Pignatti C, Marsella M, Zanforlin N. The natural history of thalassaemia intermedia. Ann N Y Acad Sci 2010; 1202: 214-20.
5.Thein S L. Genetic insights into the clinical diversity of ? thalassaemia. Br J Haematol 2004; 124: 264-74.
6.Ahmed S, Petrou M, Saleem M. Molecular genetics of -thalassaemia in Pakistan: a basis for prenatal diagnosis. Br J Haematol 1996; 94: 476-81.
7.Aessopos A, Kati M, Meletis J. Thalassaemia intermedia today: should patients regularly receive transfusions? Transfusion 2007; 47: 792-800.
8.Cappellini M D, Musallam K M, Taher A T. Insight onto the Pathophysiology and Clinical Complications of Thalassaemia Intermedia. Hemoglobin 2009; 33: 145-59.
9.Borgna-Pignatti C. Modern treatment of thalassaemia intermedia. Br J Haematol 2007; 38: 291-304.
10.Taher A T, Musallam KM, Cappellini M D, Weatherall D J. Optimal management of ? thalassaemia intermedia. Br J Haematol 2011; 152: 512-23.
Journal of the Pakistan Medical Association has agreed to receive and publish manuscripts in accordance with the principles of the following committees: